Desenvolvimento e validação de sondas in house para FISH para a síndrome de deleção 22q11.2
Luiza Emy Dorfman1,2; Patrícia Trevisan1,3; Diego A. Paskulin4; Maiara A. Floriani1; Luiza Carolina R. Scherner1; Naiane C. Bassani1;
Andressa S. Santos1; Rafael F. M. Rosa1,5; Paulo Ricardo G. Zen1,5
1.Universidade Federal de Ciências da Saúde de Porto Alegre, Porto Alegre, Rio Grande do Sul, Brazil.
2. Universidade do Vale do Rio dos Sinos, Escola de Saúde, São Leopoldo, Rio Grande do Sul, Brazil.
3. Sabin Medicina Diagnóstica, Brasília, Distrito Federal, Brazil.
4. Genex, Instituto de Exames Genéticos, Porto Alegre, Rio Grande do Sul, Brazil. 5. Irmandade Santa Casa de Misericórdia de Porto Alegre, Porto Alegre, Rio Grande do Sul, Brazil.
J Bras Patol Med Lab. 2021; 57: 1-7.
DOI: 10.5935/1676-2444.20210058
Corresponding Author
Paulo Ricardo G. Zen
ORCID: 0000-0002-7628-4877
e-mail: paulozen@ufcspa.edu.br
First submission on 03/25/21
last submission on 03/30/21
accepted for publication on 06/17/21
published on 12/20/21
ABSTRACT
Introduction: The molecular cytogenetic technology of fluorescence in situ hybridization (FISH) allows the identification of submicroscopic chromosomal alterations in congenital diseases. The 22q11.2 deletion syndrome is one of the most frequent genetic diseases in humans, and has a very broad and variable clinical phenotype, which difficults the clinical diagnosis. Although it has the possibility of submicroscopic changes identification using array-comparative genomic hybridization (CGH), multiplex ligation-dependent probe amplification (MLPA) and bacterial artificial chromosomes (BACs)-on-beads; FISH probes are still used to diagnose or confirm the results of these techniques. Objective: The present study aimed to establish the technique of manufacturing DNA probes for FISH for the diagnosis of 22q11.2 deletion with reduced costs for the public health systems. Material and methods: In house development of FISH probe assays consists of several sequential processes. Initially the BACs were identified and selected, then they were grown, and, afterwards, their DNA was extracted and amplified. The amplified DNAs were labeled with fluorochromes by Nick-translation technique. After probes development, the validation was performed on normal control samples and on altered samples. Results and conclusion: On normal samples, we achieved 93.55% of sensitivity, 99.2% of specificity and 99.8% of efficiency. These manufactured probes cost 8.5 times lower than the commercial ones.
Key words: DiGeorge syndrome; in situ hybridization fluorescence; molecular probes.
RESUMO
Introdução: A tecnologia de citogenética molecular de hibridização in situ fluorescente (FISH) permite a identificação de alterações cromossômicas submicroscópicas em doenças congênitas. A síndrome de deleção 22q11.2 é uma das doenças genéticas mais frequentes no ser humano e tem um fenótipo clínico muito amplo e variável, o que dificulta o diagnóstico clínico. Embora tenha a possibilidade de identificação de alterações submicroscópicas usando array-CGH, multiplex ligation-dependent probe amplification (MLPA) e cromossomos artificiais bacterianos (BACs)-on-beads, as sondas para FISH ainda são utilizadas para diagnosticar ou confirmar os resultados dessas técnicas. Objetivo: O objetivo deste estudo foi estabelecer a técnica de produção de sondas de ácido desoxirribonucleico (DNA) para FISH para o diagnóstico de deleção 22q11.2, com custos reduzidos para os sistemas de saúde pública. Material e métodos: O desenvolvimento interno de ensaios de sondas para FISH consiste em vários processos sequenciais. Inicialmente, os BACs foram identificados e selecionados; depois foram cultivados e, posteriormente, o seu DNA foi extraído e amplificado. Os DNAs amplificados foram marcados com fluorocromos pela técnica de Nick-translation. Após o desenvolvimento das sondas, foi efetuada a validação em amostras de controle normais e em amostras alteradas. Resultados e conclusão: Em amostras normais, conseguimos 93,55% de sensibilidade, 99,2% de especificidade e 99,8% de eficiência. Essas sondas fabricadas custaram 8,5 vezes menos do que as sondas comerciais.
Unitermos: síndrome de DiGeorge; hibridização in situ fluorescente; sondas moleculares.
RESUMEN
Introducción: La tecnología de citogenética molecular de hibridación in situ fluorescente (FISH) permite la identificación de alteraciones cromosómicas submicroscópicas en enfermedades congénitas. El síndrome de deleción 22q11.2 es una de las enfermedades genéticas más frecuentes en el ser humano y tiene un fenotipo clínico muy amplio y variable, lo que dificulta el diagnóstico clínico. Aunque es posible identificar alteraciones submicroscópicas utilizando array-CGH (hibridación genómica comparada por microarreglos), amplificación de sondas dependiente de ligandos múltiples (MLPA) y cromosomas artificiales bacterianos (BACs)-on-beads, las sondas FISH todavía se utilizan para diagnosticar o confirmar los resultados de estas técnicas. Objetivo: El objetivo de este estudio fue establecer la técnica de producción de sondas de ácido desoxirribonucleico (ADN) FISH para el diagnóstico de deleción 22q11.2, con costos reducidos para los sistemas de salud pública. Material y métodos: El desarrollo interno de ensayos de sonda para FISH consta de varios procesos secuenciales. Inicialmente, se identificaron y seleccionaron BACs; luego se cultivaron y, posteriormente, se extrajo y amplificó su ADN. Los ADN amplificados se marcaron con fluorocromos utilizando la técnica de Nick-translation. Después del desarrollo de las sondas, se realizó una validación en muestras de control normales y en muestras alteradas. Resultados y Conclusión: En muestras normales, logramos una sensibilidad del 93,55%, una especificidad del 99,2% y una eficiencia del 99,8%. Estas sondas fabricadas cuestan 8,5 veces menos que las sondas comerciales.
Palabras clave: síndrome de DiGeorge; hibridación in situ fluorescente; sondas moleculares.
INTRODUCTION
The 22q11.2 deletion syndrome presents a broad and variable clinical phenotype, characterized by facial dysmorphia, immunodeficiency, congenital cardiac alterations, palate malformations and endocrine alterations. Also known as DiGeorge syndrome (OMIM*188400) and velocardiofacial syndrome (OMIM*192430), it is associated to high rates of clinical and psychiatric comorbidities, as well as cognitive deficit(1). Moreover, it is currently considered as the most associated genetic syndrome with schizophrenia(2). It is one of the most frequent genetic diseases in humans, presenting an estimated frequency of 1 in every 3,000 to 4,000 live births(3). It is important to notice that its frequency may vary according to the studied sample and to the technique employed for detecting this alteration(4). Due to the difficulty in diagnosis, some patients only receive the syndrome confirmation during adulthood, or as part of the diagnostic follow-up of a family member more severely affected(5). The early detection of this syndrome is of fundamental importance to assure a precise diagnosis to the patients and their families, indicating the most appropriate therapy and preventing aggravations.
In spite of the possibility of chromosome identification microdeletions through array-comparative genomic hybridization (CGH), multiplex ligation-dependent probe amplification (MLPA) and bacterial artificial chromosomes (BACs)-on-Beads techniques, fluorescence in situ hybridization (FISH) probes are still being employed for diagnosis or as a results confirmation technique(6, 7). Commercial probes for the 22q11.2 deletion have a detection capability of about 85% of 3 Mb deletion cases(3) and usually consist of one probe which hybridizes on a syndrome region associated to TUPLE1 (HIRA) 22q11.2 and another one which hybridizes on a control region.
The FISH technique is used to identify the presence and location of specific regions of the genome. It employs genomic desoxyribonucleic acid (DNA) probes which are directly labeled (with fluorochromes) or indirectly (using haptens)(8). Due to several advantages regarding the karyotype, the FISH technique has gradually been seen as a complementation to the traditional chromosomes analysis. In clinical cytogenetics, FISH is applied on prenatal and postnatal diagnosis and on numeric and structural chromosomal alterations detection, on genic loci mapping and on revealing cryptic alterations(9). Because of the wide diffusion of cytogenomic techniques in the last few years, FISH has also been used for confirmation and clarification of array-CGH and single nucleotide polimorphism (SNP)-array results(10-13).
Although there is a wide range of FISH probes on the market worldwide, due to their high cost and to the possibility of innovation, manufactured homebrew probe assays are still employed(3). These kinds of probes are commonly developed from clones of BACs that consist of plasmids of Escherichia coli, which contain fragments of specific sequences of human DNA (insert) and an antibiotic resistance gene.
In Brazil, the high commercial cost of FISH probes has intensely limited its use for diagnosis in the public health system, in which this methodology is still not routinely available. Thus, mastering FISH probes development represents a technological advance for gold standard diagnosis more accessible. Because of that, it is possible to offer patients a more appropriate and direct genetic counseling, besides providing epidemiologic data collection for future studies. Apart from application of FISH methodology on diagnosis, manufacturing these probes provides a wide range of possibilities to cytogenetic research, such as the opportunity to develop new products which are not available on the market, or yet, to customize some already existent products according to our demands. Thus, this study aims to develop and validate homebrew FISH probes for 22q11.2 deletion syndrome.
MATERIAL AND METHODS
Samples
For the sensitivity, specificityand efficiency analysis of the developedprobes, we employedtwentynormal samples (negativecontrols) of healthyvolunteer donors, studentsand employees of UniversidadeFederal de Ciências da Saúde de Porto Alegre (UFCSPA). The collection of 4 ml of peripheralblood was performed in a proper collection tube. These samples wereusedonlyfordevelopedprobesvalidation, the risk to donors was onlyassociatedwith the bloodcollectionitself. The positivecontrol was obtainedfrom a sample of a patientwith 22q11.2 deletion, previouslydiagnosedusing a commercial probe, from the Hospital da Criança Santo Antônio (HCSA). No patientinformation was provided, apart fromsyndromeconfirmatoryresults, thusmaintaininganonymity. This research was approvedby the Research EthicsCommittee of bothinstitutions (2.348.732/1.871.676).
Choice of the clones
The DNA sequences or interest genes were obtained in the UCSC Genome Browser (https://genome.ucsc.edu) according to human species, hg19 assembly. For tagging of TUPLE1 (HIRA) gene on 22q11.21 region, we selected RP11-1057H19 clone and RP11-825H3 clone on 22q13.33 region (Figure 1) for control, both present in the BAC End Pairs Library, acquired from the company BACPAC Resources Center (https://bacpacresources.org/).
We created and acquired specific primers for each sequence according to what follows: RP11-1057H19,

5’-CAACATCACCCTGACACCAA-3’ (forward) and 5’-CCACCACGCCAAGCTAAT-3’ (reverse); RP11-825H3, 5’-AAACGTCTCACCGAGTTGAC-3’ (forward) and 5’-GGAGGATAAAGCAGGAGAAATGA-3’ (reverse) using a sequence of 600 bp of each clone with the PrimerQuest Tool of Integrated DNA Technologies (https://www.idtdna.com/Primerquest/Home/ Index). We selected primers which had around 20 bp each, and that generated fragments of 200 to 500 bp, with a guanine and cytosine content of 45%-55%, and melting between 54°C and 60°C (Table).
BACs cultivation
The acquired clones were cultivated overnight at 37°C in a bacteriologic incubator, in 100 ml of LB Agar 3.5% (Affymetrix, Santa Clara, CA) with chloramphenicol at 34 μg/ml. After cultivation, four to eight single isolated colonies of medium size were selected, and were put into individual cultivations containing 3 ml of LB Broth 2% (Affymetrix, Santa Clara, CA) with chloramphenicol at 34 μg/ml. The remaining material of each colony was employed for inserts verification step by polymerase chain reaction (PCR).

DNAs extraction and amplification
Only the colonies that obtained specific sequence confirmation by PCR were employed to probe development. After single colonies overnight growth in LB Broth, its DNA was extracted using QIAmp® DNA Mini Kit (Qiagen, Venlo, Netherlands), and quantified in agarose gel. The DNAs with the highest quantity and quality were selected and amplified with REPLI-g® Mini Kit (Invitrogen, Carlsbad, CA). After amplification, the DNAs were again quantified in agarose gel.
DNAs labeling and precipitation
DNAs fluorescence labeling was performed by Nick-translation (Abbott, Chicago, IL), with around 1 μg of DNA amplified. DNAs and specific reagents were incubated in Veriti 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA) during 14 h at 15°C, and, afterwards, remained for 10 min at 70°C. The RP11-1057H19 DNA was labeled with Red-dUTP (Abbott), and the RP11-825H3 DNA was labeled with Green-dUTP (Abbott). The labeled DNAs were individually mixed to Salmon Sperm® DNA (Invitrogen), Human Cot-1® DNA (Invitrogen), 3 M sodium acetate and absolute ethanol. These mixtures remained for 1 h at -85°C, after which they went through centrifugations with 70% alcohol for pellets washing. The pellets drying at room temperature. The precipitated material was dissolved in tDenHyb-2 Solution® (Insitus, Albuquerque, NM) – after this step, the probes were ready to be applied to samples on slides.
Samples culture and processing
The blood samples went through a 72 h growth in PB-MAX® Karyotyping Medium (ThermoFisher Scientific, Waltham, MA) in a CO2 incubator at 37°C. After culture, Colcemid® solution (ThermoFisher Scientific) was added to the samples for 20 min and after centrifugation supernatants materials were removed and potassium chloride (KCl) was added at 37°C for the hypotonic shock. After 15 min, the hypotonic solution action was interrupted with Carnoy’s fixative solution (3 methanol: 1 glacial acetic acid). The samples pellets were centrifuged and washed with this solution and, afterwards, were placed at -8°C and stored on slides.
Pre-treatment, hybridization and post-washing
The previously prepared slides had gone through washes with sodium citrate, pepsin solution, phosphate buffered saline solution (PBS) and formaldehyde solution. Then, they were dehydrated on increasing ethanol concentrations. Once this step was concluded, the hybridization process was initiated. For each slide, 1.5 μl of each labeled probe was mixed to 1 μl of tDenHyb-2 Solution® (Insitus), this mixture was covered by a coverslip which was sealed with rubber cement (Elmer’s, Atlanta, GA). After the cement was fully drying, the slides were incubated at 85°C for 10 min to DNAs co-denaturation. The slides materials were hybridized in a humid chamber at 37°C for 18 to 44 h. After the hybridization period, the slides were washed in sodium citrate and Tween-20 (Sigma- Aldrich, St. Louis, MO) solutions, and, afterwards, in solutions containing only sodium citrate. Following this process, DAPI counterstain (4’,6-diamidino-2-fenilindol) (Abbott) was applied and a coverslip was placed on it.
Analysis and validation
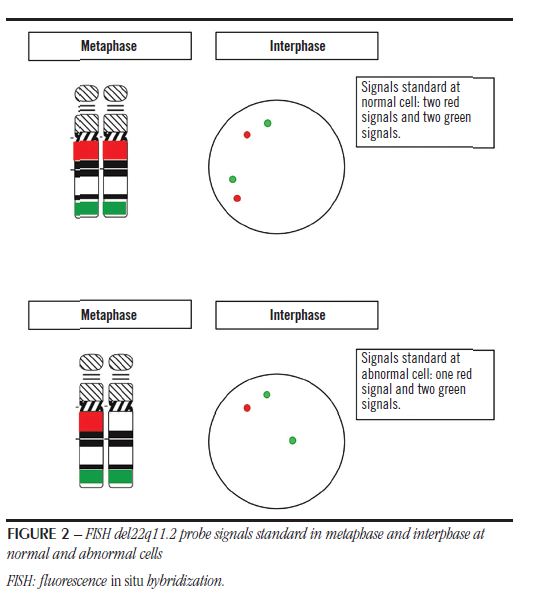
Analysis was performed on fluorescence microscope AxioImager Z2 (Zeiss, Oberkochen, Germany) and the photographic registers werecreatedusing Isis® (MetaSystems, Altlussheim, Germany). The cellcounting, performance testingandprobesvalidation criteria followed the recommendations of The AGT CytogeneticsLaboratoryManual(3), andwereperformedbytwotrainedanalysts. The cellswereevaluatedwith Green-49303 (Chroma, BellowsFalls, VT) and Orange-49305 (Chroma) filters forindividualprobesdetection. We usedtriple filter DAPI/ FITC/TexasRed-61002 (Chroma) fordevelopment set probesvisualization. Fornormalcellstwo green signalsandtwo red signalswereexpectedandtwo green andone red signal (Figure 2) for the abnormal. Analyzed data wereregisteredandusedtoperformdevelopedprobessensitivity, specificityand efficiency evaluation. To sensitivitycalculation, we counted 4,000 interphases in 20 normal samples – 200 interphases of each, considering standard green/red probe signals in each analyzed cell. To specificity calculation, we analyzed at least 100 metaphases in the 20 normal samples. To accurately guarantee the chromosomal location we used a tool from image capture Isis® (MetaSystems), called reverse DAPI, which facilitates chromosome identification based on the banding pattern adopted by conventional cytogenetics. Finally, the efficiency was according to hybridized analyzable cells percentage, among 4,000 cells counted as normal samples. In an abnormal sample, we analyzed 200 interphases and seven metaphases.
RESULTS
Ourdevelopedprobesdetected the deletion at anabnormal sample; both in interphases (Figure 3A) as well as in metaphases (Figure 4A-B). We counted 4,000 interphases in the normal samples and 3,7 86 presented the standard pattern (Figure 3B), resulting in a sensitivity of 93.55%. For each nuclei, the mean of expected signals was 187.1 (180-195). We also analyzed 125 metaphases (Figure 4C-D), among which only one did not present the standard pattern signals, thus resulting in a specificity of 99.2%. From the 4,000 analyzed interphases, 3,991 presented hybridization, showing an efficiency of 99.8%. On average, from the 200 interphases analyzed by sample, 199.5 (196-200) hybridized.

DISCUSSION AND CONCLUSION
FISH probe assays can be applied on different genomic investigation studies. Even with the development of newer and high-resolution technologies, FISH alone or in association with other techniques remains to be used in the diagnosis of congenital diseases and neoplasms(14-17). Due to high costs of commercial probes in our country, we implemented FISH probes development in our laboratory.
Besides the technique implementation, we were able to reduce the probe cost per sample to a value of U$ 8.9 for homebrew probe instead of the original cost of U$ 76 per sample. In Brazil, another factor that affects the wide use of commercial probes is delivery time, and since there is no commercial manufacturer in our country, we must import these products form foreign countries with an arrival date of around 70 days.
During the technique implementation, we also developed other probes, including a set for fusion BCR-ABL1 (breakpoint cluster region-Abelson1) genes identification. At analysis, we observed unspecific signals regarding the BCR probe. Using the reverse DAPI tool, we observed that signals always appeared on the same chromosomes, and thus, using basic local alignment search tool (BLAST tool) (https://blast.ncbi.nlm.nih.gov/ Blast.cgi), we compared the clone sequence with other human sequences for verifying its specificity regarding the interest sequence. We confirmed the sequences compatibility of the clone with regions other than chromosomes 22. Therefore, we strongly suggest the inclusion of the clone sequence verification step in the BLAST tool prior to purchase of the clone. This step should be included in the protocols of any study that uses BACs that require similarity.
The next goals of our research group are the development of probes designed for the diagnosis of other chromosomal microdeletion syndromes such as Prader-Willi/Angelman, Williams- Beuren and also for neoplasms diagnosis, including inv(16)
(p13.1q22), t(15;17)(q22;q12) that are frequent in acute myeloid leukemia.
ACKNOWLEDGEMENT
We would like to thank Professor Dr. Marileila Varella-Garcia (Cytogenetics Core, Cancer Center, University of Colorado, Aurora) for sharing the protocols for probes development, and the Professor Dr. Mariluce Riegel (Laboratório de Citogenética Molecular, Hospital de Clínicas de Porto Alegre) who provide us FISH’s pretreatment and post-wash protocols.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
FUNDING SOURCES
Supported by Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS), (1213-2551/13.0); Support Program for University Extension of Ministry of Education and Culture (PROEXT); National Council for Scientific and Technological Development (CNPq) and Coordination of Superior Level Staff Improvement (CAPES). Research productivity fellowship Brazil (CNPq) (301834/2016-4).
AUTHOR CONTRIBUTIONS
All authors contributed to data analysis, drafting and revising the article. All the authors gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
REFERENCES
- Verheij E, Speleman L, van der Molen ABM, Thomeer HG. Congenital respiratory tract disorders in 22q11.2 deletion syndrome. Int J Pediatr Otorhinolaryngol. 2018, 104: 1-4.
- Weinberger R, Weisman O, Guri Y, Harel T, Weizman A, Gothelf D. The interaction between neurocognitive functioning, subthreshold psychotic symptoms and pharmacotherapy in 22q11.2 deletion syndrome: a longitudinal comparative study. Eur Psychiatry. 2018, 48(1): 20-26.
- Arsham MS, Barch MJ, Lawce HJ. The AGT cytogenetics laboratory manual. 4th ed. John Wiley & Sons; 2017.
- Belangero SIN, Bellucco FT, Kulikowski LD, Christofolini DM, Cernach MC, Melaragno MI. Deleção 22q11.2 em pacientes com defeito cardíaco conotruncal e fenótipo da síndrome da deleção 22q11. 2. Arq Bras Cardiol. 2009; 92(4): 307-11.
- Benn P, Iyengar S, Crowley TB, et al. Pediatric healthcare costs for patients with 22q11.2 deletion syndrome. Mol Genet Genomic Med. 2017; 5(6): 631-38.
- Grati FR, Gomes DM, Ferreira JCPB, et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn. 2015; 35(8): 801-09.
- Poirsier C, Besseau-AyasseJ, Schluth-Bolard C, et al. A French multicenterstudy of over 700 patientswith 22q11 deletionsdiagnosedusing FISH or aCGH. EurJ Hum Genet. 2016; 24(6): 844-51.
- Liehr T. Fluorescence in situ hybridization (FISH). Springer Berlin Heidelberg; 2017.
- Wan TS, Ma ES. Molecular cytogenetics: an indispensable tool for cancer diagnosis. Chang Gung Med J. 2012; 35(2): 96-110.
- Akalin I, Bozdag S, Spielmann M, Basaran SY, Nanda I, Klopocki E. Partial trisomy 1q41‐qter and partial trisomy 9pter‐9q21. 32 in a newborn infant: an array CGH analysis and review. Am J Med Genet A. 2014; 164(2): 490-94.
- Torti EE, Braddock SR, Bernreuter K, Batanian JR. Oculo‐auriculo‐vertebral spectrum, cat eye, and distal 22q11 microdeletion syndromes: a unique double rearrangement. Am J Med Genet A. 2013; 161(8): 1992-98.
- Rosenfeld JA, Patel A. Advances in pediatric genetic tests: chromosomal microarrays: understanding genetics of neurodevelopmental disorders and congenital anomalies. J Pediatr Genet. 2017; 6(1): 42.
- Pace NP, Maggouta F, Twigden M, Borg I. Molecular cytogenetic characterisation of a novel de novo ring chromosome 6 involving a terminal 6p deletion and terminal 6q duplication in the different arms of the same chromosome. Mol Cytogenet. 2017; 10(1): 1-6.
- Hatano M, Fukuzawa R, Hasegawa Y. The mosaicism ratio of 45, X may explain the phenotype in a case of mixed gonadal dysgenesis. Sex Dev. 2018; 12(4): 175-79.
- Luo A, Cheng D, Yuan S, et al. Maternal interchromosomal insertional translocation leading to 1q43-q44 deletion and duplication in two siblings. Mol Cytogenet. 2018; 11(1): 1-8.
- Panagopoulos I, Gorunova L, Jacobsen EM, Andersen K, Micci F, Heim S. RUNX1-PDCD6 fusion resulting from a novel t (5; 21)(p15; q22) chromosome translocation in myelodysplastic syndrome secondary to chronic lymphocytic leukemia. PloS One. 2018; 13(4): e0196181.
- Wemmert S, Linxweiler M, Lerner C, et al. Combinational chromosomal aneuploidies and HPV status for prediction of head and neck squamous cell carcinoma prognosis in biopsies and cytological preparations. J Cancer Res Clin Oncol. 2018, 144(6): 1129-41.

